
Genomics Reporting Implementation Guide
2.0.0 - trial-use
Genomics Reporting Implementation Guide
2.0.0 - trial-use
This page is part of the Genetic Reporting Implementation Guide (v2.0.0: STU 2) based on FHIR (HL7® FHIR® Standard) R4. The current version which supersedes this version is 3.0.0. For a full list of available versions, see the Directory of published versions
Contents:
This page defines the core profiles and concepts that would be expected to be present in most genomic reports, regardless of type, and how those profiles relate to each other. Concepts covered include the genomics report itself and the high-level categories of observations and other elements that make up the report, such as patient, specimen, variants, haplotypes, genotypes, etc.
This table describes the categories of data contained in this implementation guide.
| Genomics Report | Groups together all the structured data being reported for a genomic testing. |
|---|---|
| Overall Interpretations | Reported when variant analysis (sequencing or targeted variants) is done. Provides a coarse overall interpretation of the results reported. |
| Genomic Findings | These are observations about the specimen's genomic characteristics. For example, a chromosomal abnormality, genotype, haplotype, or variant that was detected. |
| Genomic Implications | These represent observations where the Observation.subject is typically the Patient and the Observation.derivedFrom should refer to Genomic Findings. For example, "Patient may have increased susceptibility to heart attacks" |
| Region Studied | These are observations describing the region or regions that were studied as part of this Genomics Report. |
| Other Observations | The results of tests other than sequenced genomic variants may also be included the report. |
| Recommended Actions | Specific actions be taken, such as genomic counseling, re-testing, adjusting drug dosages, etc. - driven by the results found. |
| Contextual Resources | Other resources that provide contextual details. |
The genomics report is the focus of all genomic reporting. It conveys metadata about the overall report (what kind of report it was, when it was written, who wrote it, final vs. draft, etc.). It also typically includes a rendered version for review by a clinician. It also groups together all relevant information found as part of the genomic analysis (Rules for relevancy will depend on the type of testing ordered, the reason for testing and the policies of the lab). Most of the structured genomic information is expressed as FHIR Observations. Any recommendations that come with the report are expressed as FHIR Tasks. The report can be organized into sub-reports using core DiagnosticReport extensions like extends or summaryOf, which is especially useful for later analysis steps. Additionally, an observation can be used to group content for viewing purposes or to indicate a higher-level panel (with a specific LOINC panel code in the Observation.code for example).
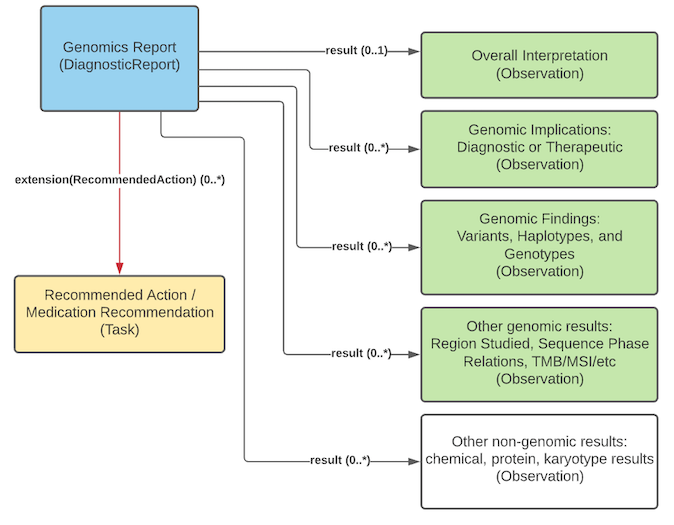
Genomic Report Overview
Genomics Report, Overall Interpretation, Genomic Findings, Genomic Implications, Region Studied, Recommended Actions
Again, if needed, large or complex genomic reports may be broken down into sub-reports using core DiagnosticReport extensions like extends or summaryOf. This approach is particularly useful when different labs or services are performing later steps in the analysis, for example. Or a panel Observation.code can be used.
In some cases, the lab or other reporting organization may generate risk assessments as part of their reports. These are referenced from a report or an observation from the Genomics Risk extension.
Results observation profiles, like genomic observations, are typically referenced directly by a Diagnostic Report. For genomics this would be in the genomics report. The genetic findings, implications and region studied profiles all contain links to computably define their composite relationships (e.g., the variant observation is referenced within the implication profile using derivedFrom). However, observations could be organized into groups by other observations. See this grouping guidance for an overview with examples and considerations for processing reports. Be aware that consumers of Genomic Diagnostic Report MUST navigate through all hasMember relations and navigate through derivedFrom relationships to ensure processing of all clinically relevant information.
In a future release, the genomic file guidance below will be changed to utilize updates coming to the
DiagnosticReport.mediabackbone element. In the FHIR R5 release, the attribute will refer toDocumentReferenceresource instead of aMediaresource, and the corresponding guidance will be updated to better support the use case here. The guidance below for file attachments needs to be carefully reviewed and considered. There are potential issues, and the Work Group welcomes feedback.
While this implementation guide strives to provide structured genomic data via a variety of observations, there are use cases that warrant a deeper level of data than this guide allows. These use cases might be best served by sending commonly used files (VCF, BAM, CRAM, MAF, BED to name a few) with the report. To promote a consistent approach to sharing these files, we provide the guidance in this section. Even with this guidance, note that best practices in exchanging these files along with the metadata necessary to make use of them through a FHIR API, remains a complicated and open issue. See operations for a description of an experimental alternative workflow.
When sending these genomic files, implementers should utilize the GenomicsDocumentReference profile to send the file content along with meaningful metadata. This approach allows the files to have their own existence and enables queries to find them via the DocumentReference resource. To enable linking from the report to files, the Genomics File extension can be used to reference files from the GenomicsReport profile. This can be helpful for those processing the report to easily identify the files that were sent along with the report.
When sending genomic files there are many considerations. For example, it is not unusual for files to be gigabytes in size. The DocumentReference resource has different options to evaluate for your use case. If embedded directly using DocumentReference.content.attachment.data, servers receiving the files may have size constraints per resource or per transaction which may limit your options. Instead of sending a large data file, the file can be referenced by a URL and title using the using DocumentReference.content.attachment.url element. This can point to an online resource that hosts the file or from where the file can be accessed. For genomic files, the host is likely not the FHIR server providing the DocumentReference data instance. Be aware that use of DocumentReference to provide access to files through URLs introduces authorization requirements that are out of scope of this Implementation Guide. With either of these approaches, it is important to note the files might be compressed. Also, the mime types for these genomic files will vary, or possibly be missing. The senders of the data should include as much metadata as possible to enable the receiver to appropriately handle the files.
For receivers to make use of these files, many facets of the generation of the files will be needed, such as what pipelines, tools, and settings were used. The intended downstream use cases must be carefully evaluated to ensure appropriate file preparation. The DocumentReference.description might be helpful for a sending system to provide guidance on how the file was generated. A fully computable approach for this issue has yet to be defined.
It should be noted that the Genomic File extension is not an appropriate way to send a copy of the report (e.g., PDF or other document containing the written report). Instead, use DiagnosticReport.presentedForm. Also note the Genomic File extension is different than the Genomics Artifact extension, which is used to reference citations, evidence and other supporting documentation for the observation or report. Another approach which should be avoided (at least for this current release) is the DiagnosticReport.media attribute. Its definition focuses on "Key images associated with this report" which does not align well with this use case. As noted in the STU note above, this will be changing in a future release.
A full, detailed implementation discussion is outside the scope of this IG. In a future release, this IG may include other profiles or artifacts along with more specific guidance.
Observations are the core representation of structured genomic information. This guide defines a number of Observation profiles, with common underlying components and constraints being inherited from abstract profiles as shown in the following diagram. The profiles and their specific usage will be defined in more detail below and on the other pages of this guide.
All genomic observations are derived from a common abstract profile that asserts they should have a category, effective date, issued date and status.

Genomic Observations
Genomics Base, Overall Interpretation, Genomic Finding (abstract), Variant, Haplotype, Genotype, Region Studied, Sequence Phase Relationship, Genomic Implication (abstract), Therapeutic Implication, Diagnostic Implication
Overall interpretation is a high-level summary observation that applies to the whole report. Their purpose is to answer the question "Did you find anything when you did the test I asked you to do?"
Overall Interpretation is what the laboratory declares as the summary result of the test (e.g., Positive, Negative, Unknown) and is typically used when the genomic test was looking for a particular genomically-based disease. It allows indication of whether genomic results known to be associated with the disease was found or not.
The primary focus of genomic testing is making Genomic Findings. These are the fine and/or coarse-grained descriptions of a specimen's genomic characteristics. It is this information that leads to the Overall Interpretations for the report, as well as Genomic Implications that are used to convey the potential impact of the genomic findings for the subject of the test.
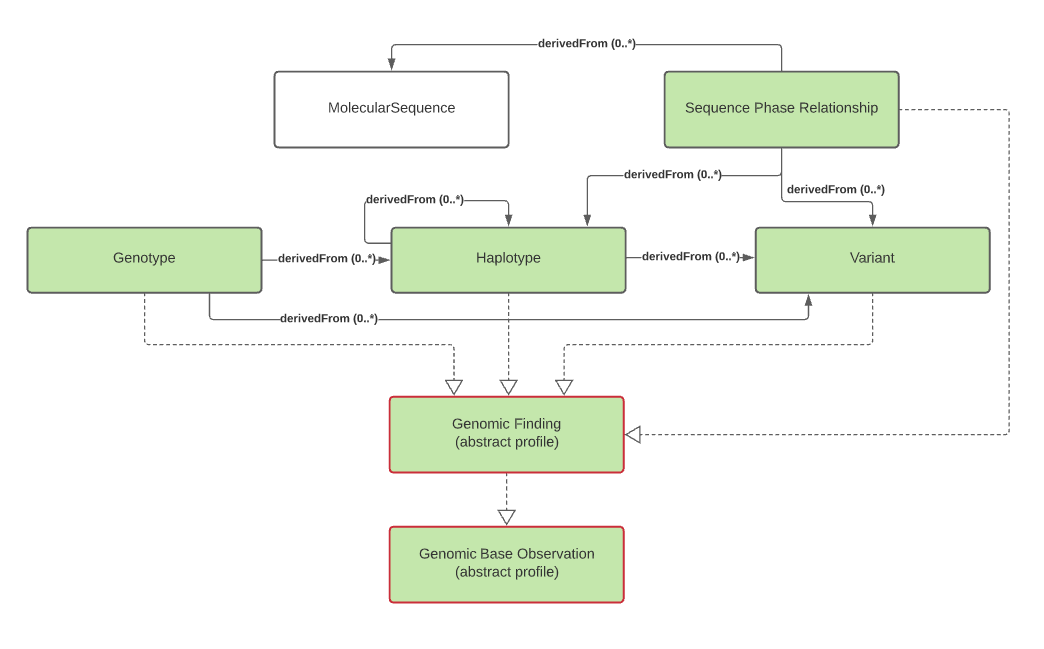
Genomic Findings
Genomic Finding, Genotype, Haplotype, Variant (or see Variant Reporting), Sequence Phase Relationship
Genomic Findings can be subdivided into types of findings:
These categories of observations have relationships. Haplotypes can be identified based on the presence of variants. Genotypes can be identified based on the presence of haplotypes and/or variants. All three can be expressed as a combination of one or more sequences.
Genotype is used to convey corresponding haplotypes or variations at a particular locus. Many genotypes are expressed as simple strings, and can be conveyed in genotype.valueCodeableConcept.text. In some cases, genotypes are sufficiently standardized to be conveyed as codes in genotype.valueCodeableConcept.code.
TPMT *1/*3A represents the TPMT *1 haplotype (or 'star allele') on one chromosome and the TPMT *3A haplotype on the homologous chromosomeA/C represents a heterozygous "A" and a heterozygous "C" at SNP rs1142345. For HLA, KIR, and other genes in the immunogenomics domain, the National Marrow Donor Program (NMDP) led a community effort to define the Genotype List String (GL String) grammar, described here. Notably, the GL String uses '+' as a delimiter between alleles in a genotype. It also has delimiters for ambiguous genotypes, allele lists, and haplotypes.
For Pharmacogenomics, we define here a simple grammar of [HGNC gene symbol followed by white space followed by a slash ('/') delimited list of haplotype codes], where the codeSystem is set to the codeSystem of the haplotypes (e.g., PharmVar).
Here are some examples that are standardized, note that there are some other examples which still lack standardization today.
{
"valueCodeableConcept": {
"text": "A/C at SNP rs1142345"
}
}
{
"valueCodeableConcept": {
"coding": [{
"system": "http://glstring.org",
"version": "1.0",
"code": "#hla#3.23#HLA-A*01:01:01:01/HLA-A*01:02+HLA-A*24:02:01:01"
}]
}
}
{
"valueCodeableConcept": {
"coding": [{
"system": "https://www.pharmvar.org/",
"code": "PV00044",
"display": "CYP2C9*2.001"
}]
}
}
At present, implications are noted as explicit observations about the patient/subject. However, it's not clear this is the correct approach. The work group is evaluating introducing a new resource that allows conveying "knowledge" about a variant in a patient-independent way. This would allow saying "this variant is associated with an increased risk of cardiovascular disease" rather than "based on this variant, the patient is at an increased risk of cardiovascular disease", which isn't necessarily a determination the reporting organization may wish to assert. Feedback is welcome.
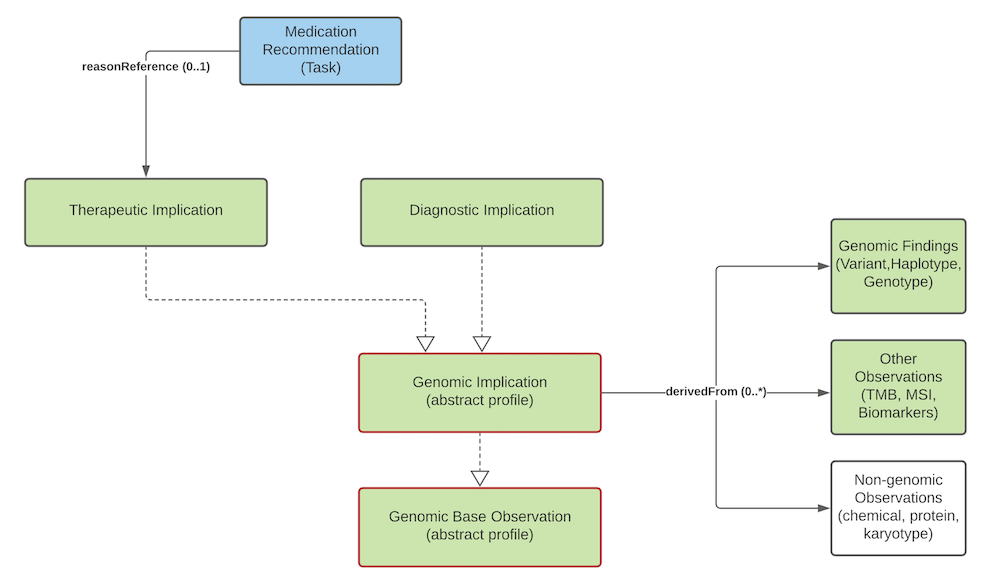
Genomic Implications
Genomic Implication, Therapeutic Implication, Diagnostic Implication
Genomic Implications are assertions of likely effects genomic results on the patient, tumor, or other subject. Implications are relevant for areas of genomic testing including inherited disease, pharmacogenomics and somatic. For inherited diseases, a diagnostic implication indicates the likelihood of inheritance of a particular disease (the associated-phenotype) as well as how inheritance is likely to occur (mode-of-inheritance). For other more specific guidance, see the pharmacogenomics and somatics pages.
Indicating AND and OR:
There are several cases where it is necessary to differentiate 'AND' conditions (e.g. both drug X AND drug Y, in combination, are indicated in the presence of a variant) vs. 'OR' conditions (e.g., either drug X OR drug Y are indicated in the presence of a variant). This situation is not unique to genomic implications, and arises elsewhere within FHIR (e.g. FHIR Search) and outside of FHIR (e.g., ClinVar submission API condition set). To be consistent with other precedents: Where Diagnostic and Therapeutic implications have fields with cardinality >1, the inclusion of multiple values within a field shall indicate an 'AND' condition. An 'OR' condition is represented by multiple observation instances. See note.Implications inherit a common set of elements:
genomics-artifact supports conveying references to citations, supporting documentation and other information relevant to the asserted implicationevidence-level (component) indicates the strength of the evidence behind the asserted implicationprognosis (component) indicates the likely diagnostic or therapeutic outcome of the implicationHere are some examples of how the evidence-level component can be used (NOTE - they would be used in the appropriate, use case specific profile):
{
"valueCodeableConcept": {
"coding": [{
"system": "https://www.acmg.org",
"code": "PS1",
},
{
"system": "https://www.acmg.org",
"code": "PM2"
}
]
}
}
{
"valueCodeableConcept": {
"coding": [{
"system": "https://www.pharmgkb.org/",
"code": "Level 1A",
}
]
}
}
{
"valueCodeableConcept": {
"coding": [{
"system": "https://www.clinicalgenome.org/mvld/",
"code": "Tier1 LevelA",
}
]
}
}
The Region Studied profile is used to assert actual regions studied for the performed test(s). Intended coverage areas may differ from actual coverage areas (e.g., due to technical limitations during test performance). It can assert and describe actual regions studied for the performed test(s). It can provide this description at the known level of detail using components.
The results of tests other than genomic findings may also be included the report. One example is the Tumor Mutation Burden profile, which captures the total number of mutations (changes) found in the DNA of cancer cells. Another example is Microsatellite Instability, a condition of genetic hypermutability that results from impaired DNA mismatch repair.
These actions can be specific recommendations be taken and are driven by the results found. These can be a Follow-up Recommendation which can be used to indicate when some sort of follow-up (additional testing, genetic counseling) is required, or a Medication Recommendation which can be used to propose medication recommendations based on the results of the test.
There are a number of resources that are used within this guide to provide additional data as both input and output of the test.
The ServiceRequest resource typically represents a clinician order. It can also represent a lab-side filler order, a reflex order or even a plan or recommendation. These uses are distinguished via the intent element. The primary test to perform is captured in ServiceRequest.code. However, qualifications on what variants, medications, diseases, and other aspects to search on can be conveyed using the orderDetail element. The service requests and the reports resulting from them can be associated to patients, to specimens or both.
Orders can point to other sources of information used to support the analysis performed as part of genomic testing. Genomic reports can refer to this the information that was considered as part of the report - whether provided as part of the order or made available subsequently by the patient or clinicians or otherwise retrieved. The figure above shows these relationships through the extension SupportingInformation, which can be to any resource but would typically reference Observations, FamilyMemberHistory records (including records that comply with Family member history for genomics analysis and RiskAssessments.
To allow searching and appropriate navigation, the diagnostic report, observations, and tasks must be able to stand on their own. They need to be related to the associated patient and/or specimen, the order that initiated the testing, the lab that performed the testing, etc. FHIR design principles dictate that these associations be present on every resource instance. That's because each resource could be accessed on its own as part of a query response, embedded in a document or message, passed to a decision support engine, etc. However, this is still relatively lightweight because the information is included by reference only.
The following diagram shows the relationships between the diagnostic report, observations, and other elements used in the profile. Note that there is no expectation that all relationships will point to the same instances. In special cases, a genomic report may involve multiple patients or multiple specimens. As mentioned, the extends, summaryOf can be used well to provide additional organization.
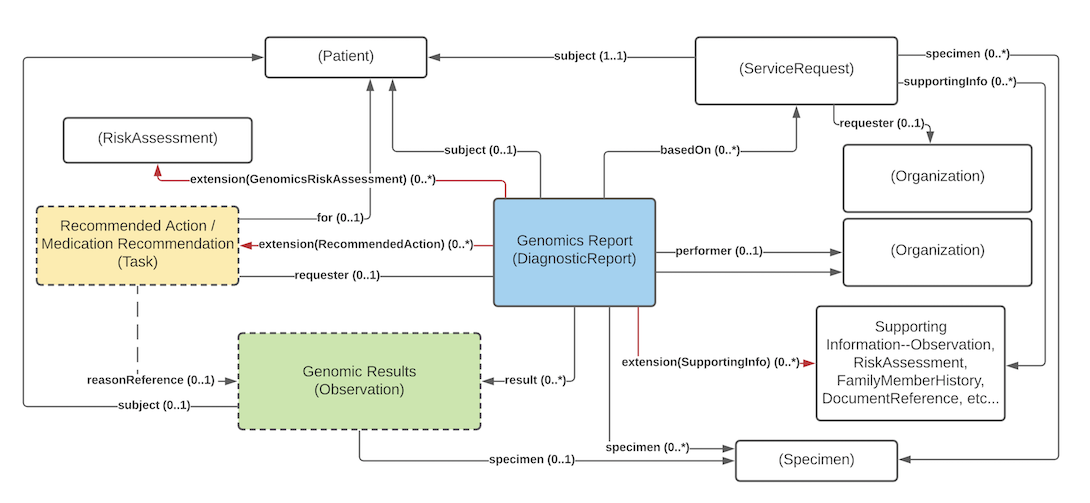
Key points to take from this diagram:
requisition identifier.Observation.derivedFrom attribute. For example, an interpretation that "deletions or duplication were found" might be supported by observations of variants that contain deletions and/or duplication.Observation.derivedFrom attribute. For example, in a genomic report, it's not acceptable to imply "patient is an increased metabolizer of drug X" without also indicating the variant, haplotype or genotype found that supports that implication.The following is derived from the CORE FHIR guidance on Diagnostic Report Resource and Observation Resource
For receivers of genetic reports, to ensure all clinically relevant information is processed consumers of Genomic Diagnostic reports MUST navigate through all hasMember relations, and navigate through derivedFrom when processing Genomic Diagnostic reports.
For senders of Genomic Diagnostic reports, it is up to receiving systems to arrange/use the genomic observations appropriately. However, some systems may only process just the first layer of a DiagnosticReport.results. Thus, when sending the report and results, we recommend including result links to all the genomic observation results being returned. For more complex reporting use cases, consider using the summaryOf extension on DiagnosticReport for creating meaningful subsets of observations.
As a note on querying, the relationships indicated by hasMember, derivedFrom and the computable meaning conveyed by observation.code and component.code are critical for specificity in querying and getting comprehensive search results. Additionally, values found in elements like component.value and observation.value are useful as well. However, the meanings of the values are predicated on by the component.code or observation.code to which they associate. There is a page in this IG with query guidance that show how to use components, relationships (references) and the iterative hasMember to find results. The use of a grouping observation does not influence the ability to query. For example, the profile of a variant contains a value for the gene name component. Querying for all variants for the patient in a given gene can be done through the gene name component.
This Implementation Guide uses FHIR profiling to define constraints on base resources to provide an interoperable structure for genomic concepts. For example, the Variant profile defines the structure for delivering variant attributes. While these profiles seek to fully describe each concept, there might be other attributes that need to be delivered. While we suggest some caution in doing this, as it makes implementations less consistent and therefore less interoperable, there are at least two ways this can be accomplished:
| Custom extensions | Implementers can use the extensibility features built into FHIR. This guide introduces a number of extensions, but other use case specific extensions can be introduced when needed. |
|---|---|
| Open slicing | When constraining attributes that can contain multiple values, the FHIR concept of slicing is used. The slices we define are all 'open' which means that while the specific rules should be used when possible, implementers are free to send additional data. For example, in Observation.code, our profiles require a specific code be sent, but still allow other codes to be delivered. Another example is in Observation.component where additional attributes can be described in Observation.component.code and their value delivered in Observation.component.value[x]. |
The work group is actively requesting feedback from the community on additional codes, values, and extensions that stakeholders have found helpful when implementing the structures and use cases defined in this guide.
IG © 2022+ HL7 Clinical Genomics Working Group. Package hl7.fhir.uv.genomics-reporting#2.0.0 based on FHIR 4.0.1. Generated 2022-05-09
Links: Table of Contents |
QA Report
| Version History |
Search |
 |
Propose a change
|
Propose a change