Release 5 Draft Ballot

 Diagnostics
DiagnosticsThis page is part of the FHIR Specification (v4.6.0: R5 Draft Ballot - see ballot notes). The current version which supercedes this version is 5.0.0. For a full list of available versions, see the Directory of published versions  . Page versions: R5 R4B R4 R3
. Page versions: R5 R4B R4 R3
| Clinical Genomics Work Group | Maturity Level: 1 | Standards Status: Trial Use |
Table of Contents
Trial-Use Note:
The material on this page is currently undergoing work to be refactored in a future release as further analysis is done, for example as part of the Genomics Reporting Implementation Guide
Rapid advances in sequencing technologies, clinical genetics tests for whole genome and exome sequencing are allowing sophisticated genetics testing to be used by providers and patients in clinical decisions. Results from such tests are used to identify distinct genetic variants that may contribute to syndromes, conditions and/or predictive responses to treatments. The implementation of precision medicine will depend upon having such data to diagnose patients, choose medications, and predict the course of disease and care, but will require standards and effective user interfaces.
A current technical challenge exists in interoperability, the ability to access and share clinical and genetics data. The challenges of interoperability includes collection, coding, and retrieval to scale. An individual's genetic data set is large, complex and requires curation. Unfortunately, incompatible systems and nomenclatures are already in use. A standards-based ontology that could be adopted to integrate both genetic data and clinical information systems will be crucial to accelerating the integration of precision medicine and to make sense of genetic testing results in a complete clinical context.
One approach for collecting, coding, and retrieving genetics data comes
from the Global Alliance for Genomics and Health
(GA4GH). The
GA4GH organization has built and is refining an API and data
model for the exchange of full sequence genomic information across multiple
research organizations and platforms. The GA4GH focuses on the needs of
researchers.
A second approach is evolving from HL7 through FHIR. FHIR is attractive because it is relatively easy to implement because it is comprised of a set of modular components called resources, which can be easily and incrementally assembled into working systems. The clinical requirements for genetics data is, relative to genomics research needs, utilitarian and reductive because it is about distilling and extracting particular genetics data produced by ever more sophisticated testing for use at the point-of-care. This has made FHIR a very functional framework to initiate an interoperable clinical genetics data standardization to which multiple stakeholders have contributed to this guide.
FHIR DSTU2 introduced a standard genetics profile that applies to the FHIR Observation resource. Using this profile, Observation payloads can return genetic testing results in a standardized manner. In STU3, a new resource to be called MolecularSequence was created allowing for increased granularity and less ambiguity.
In addition to the MolecularSequence, a new FHIR Implementation Guide Genomics Reporting Implementation Guide is being developed. At present, this implementation guide focuses solely on data structures - what data should be/might be present and how it should be organized. It does not address workflows around how reports are requested, created, approved, routed, delivered, amended, etc. The implementation guide is also paradigm-independent - the data structures presented here could be used in RESTful, messaging, documents or other mechanisms.
This resource will be used to hold clinically relevant sequence data in a manner that is both efficient and versatile integrating new and as yet undefined types of genomic and other -omics data that will soon be commonly entered into health records for clinical use. MolecularSequence will be leveraged by other FHIR resources, including Observation. This is consistent with how all FHIR resources are designed and used.
With this resource and profiles found in the Genomics Reporting Implementation Guide , FHIR can support a large set of clinical use cases (see Section 9, 10 , and 11) and is thus positioned to address all emergent -omics use cases, including Next-Generation Sequencing (NGS).
These tools are simple to implement, will optimize payload sizes, and help developers avoid redundant retrieval of data.
The September 2014 Informative Ballot (“HL7 Clinical Genomics, Domain Analysis Model: Clinical Sequencing Release 1”) provided guiding use cases,
which initially informed development of the initial Standard Genetics profile that is found in FHIR DSTU2. The same use cases also led to a
second Project to develop a MolecularSequence resource (“Develop FHIR sequence resource for Clinical Genomics”). A preliminary effort to address these issues has been explored and published in context of the Substitutable Medical Applications and Reusable Technologies (SMART) Platforms Project and described in an article (“SMART on FHIR Genomics: Facilitating standardized clinico-genomic apps ”).
| Name | Flags | Card. | Type | Description & Constraints |
|---|---|---|---|---|
| TU | DomainResource | Information about a biological sequence + Rule: Only 0 and 1 are valid for coordinateSystem Elements defined in Ancestors: id, meta, implicitRules, language, text, contained, extension, modifierExtension | ||
 | Σ | 0..* | Identifier | Unique ID for this particular sequence. This is a FHIR-defined id |
| Σ | 0..1 | code | aa | dna | rna sequenceType (Required) |
| Σ | 1..1 | integer | Base number of coordinate system (0 for 0-based numbering or coordinates, inclusive start, exclusive end, 1 for 1-based numbering, inclusive start, inclusive end) |
| Σ | 0..1 | Reference(Patient) | Who and/or what this is about |
| Σ | 0..1 | Reference(Specimen) | Specimen used for sequencing |
| Σ | 0..1 | Reference(Device) | The method for sequencing |
| Σ | 0..1 | Reference(Organization) | Who should be responsible for test result |
| Σ | 0..1 | Quantity | The number of copies of the sequence of interest. (RNASeq) |
 | ΣI | 0..1 | BackboneElement | A sequence used as reference + Rule: GenomeBuild and chromosome must be both contained if either one of them is contained + Rule: Have and only have one of the following elements in referenceSeq : 1. genomeBuild ; 2 referenceSeqId; 3. referenceSeqPointer; 4. referenceSeqString; |
 | Σ | 0..1 | CodeableConcept | Chromosome containing genetic finding chromosome-human (Example) |
| Σ | 0..1 | string | The Genome Build used for reference, following GRCh build versions e.g. 'GRCh 37' |
| Σ | 0..1 | code | sense | antisense orientationType (Required) |
| Σ | 0..1 | CodeableConcept | Reference identifier ENSEMBL (Example) |
| Σ | 0..1 | Reference(MolecularSequence) | A pointer to another MolecularSequence entity as reference sequence |
| Σ | 0..1 | string | A string to represent reference sequence |
| Σ | 0..1 | code | watson | crick strandType (Required) |
| Σ | 0..1 | integer | Start position of the window on the reference sequence |
 | Σ | 0..1 | integer | End position of the window on the reference sequence |
| Σ | 0..* | BackboneElement | Variant in sequence |
| Σ | 0..1 | integer | Start position of the variant on the reference sequence |
| Σ | 0..1 | integer | End position of the variant on the reference sequence |
| Σ | 0..1 | string | Allele that was observed |
| Σ | 0..1 | string | Allele in the reference sequence |
| Σ | 0..1 | string | Extended CIGAR string for aligning the sequence with reference bases |
| Σ | 0..1 | Reference(Observation) | Pointer to observed variant information |
| Σ | 0..1 | string | Sequence that was observed |
| Σ | 0..* | BackboneElement | An set of value as quality of sequence |
| Σ | 1..1 | code | indel | snp | unknown qualityType (Required) |
| Σ | 0..1 | CodeableConcept | Standard sequence for comparison FDA-StandardSequence (Example) |
| Σ | 0..1 | integer | Start position of the sequence |
| Σ | 0..1 | integer | End position of the sequence |
| Σ | 0..1 | Quantity | Quality score for the comparison |
| Σ | 0..1 | CodeableConcept | Method to get quality FDA-Method (Example) |
| Σ | 0..1 | decimal | True positives from the perspective of the truth data |
| Σ | 0..1 | decimal | True positives from the perspective of the query data |
| Σ | 0..1 | decimal | False negatives |
| Σ | 0..1 | decimal | False positives |
| Σ | 0..1 | decimal | False positives where the non-REF alleles in the Truth and Query Call Sets match |
| Σ | 0..1 | decimal | Precision of comparison |
| Σ | 0..1 | decimal | Recall of comparison |
| Σ | 0..1 | decimal | F-score |
 | Σ | 0..1 | BackboneElement | Receiver Operator Characteristic (ROC) Curve |
| Σ | 0..* | integer | Genotype quality score |
| Σ | 0..* | integer | Roc score true positive numbers |
| Σ | 0..* | integer | Roc score false positive numbers |
| Σ | 0..* | integer | Roc score false negative numbers |
| Σ | 0..* | decimal | Precision of the GQ score |
| Σ | 0..* | decimal | Sensitivity of the GQ score |
| Σ | 0..* | decimal | FScore of the GQ score |
| Σ | 0..1 | integer | Average number of reads representing a given nucleotide in the reconstructed sequence |
| Σ | 0..* | BackboneElement | External repository which contains detailed report related with observedSeq in this resource |
| Σ | 1..1 | code | directlink | openapi | login | oauth | other repositoryType (Required) |
| Σ | 0..1 | uri | URI of the repository |
| Σ | 0..1 | string | Repository's name |
| Σ | 0..1 | string | Id of the dataset that used to call for dataset in repository |
| Σ | 0..1 | string | Id of the variantset that used to call for variantset in repository |
| Σ | 0..1 | string | Id of the read |
| Σ | 0..* | Reference(MolecularSequence) | Pointer to next atomic sequence |
| Σ | 0..* | BackboneElement | Structural variant |
| Σ | 0..1 | CodeableConcept | Structural variant change type LOINC LL379-9 answerlist (Required) |
| Σ | 0..1 | boolean | Does the structural variant have base pair resolution breakpoints? |
| Σ | 0..1 | integer | Structural variant length |
| Σ | 0..1 | BackboneElement | Structural variant outer |
| Σ | 0..1 | integer | Structural variant outer start |
| Σ | 0..1 | integer | Structural variant outer end |
| Σ | 0..1 | BackboneElement | Structural variant inner |
| Σ | 0..1 | integer | Structural variant inner start |
| Σ | 0..1 | integer | Structural variant inner end |
| Documentation for this format | ||||
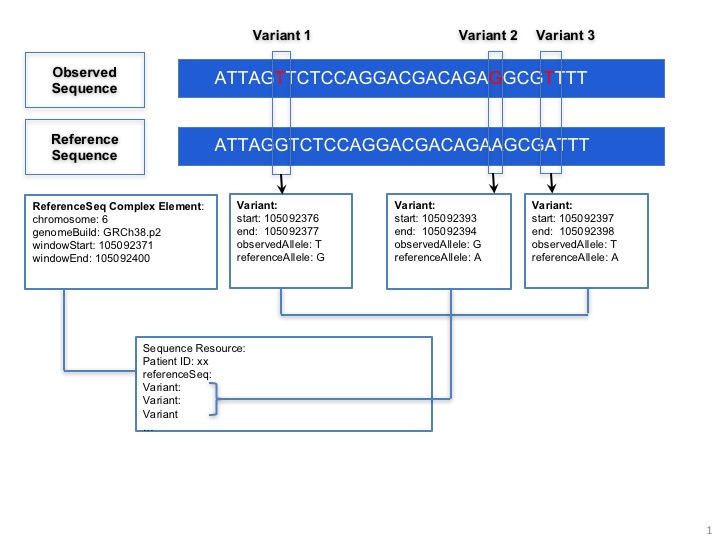
The MolecularSequence resource is designed for next-generation sequencing data. Patients’ observed sequences should be represented by recording reference sequence id/string and detected variants. To specify how it proceed, here is a picture below:
MolecularSequence.coordinateSystem: This element shall be constrained into only two possible values: 0 for 0-based system and 1 for 1-based system. Below is the picture that could explain what’s the difference between these two systems:
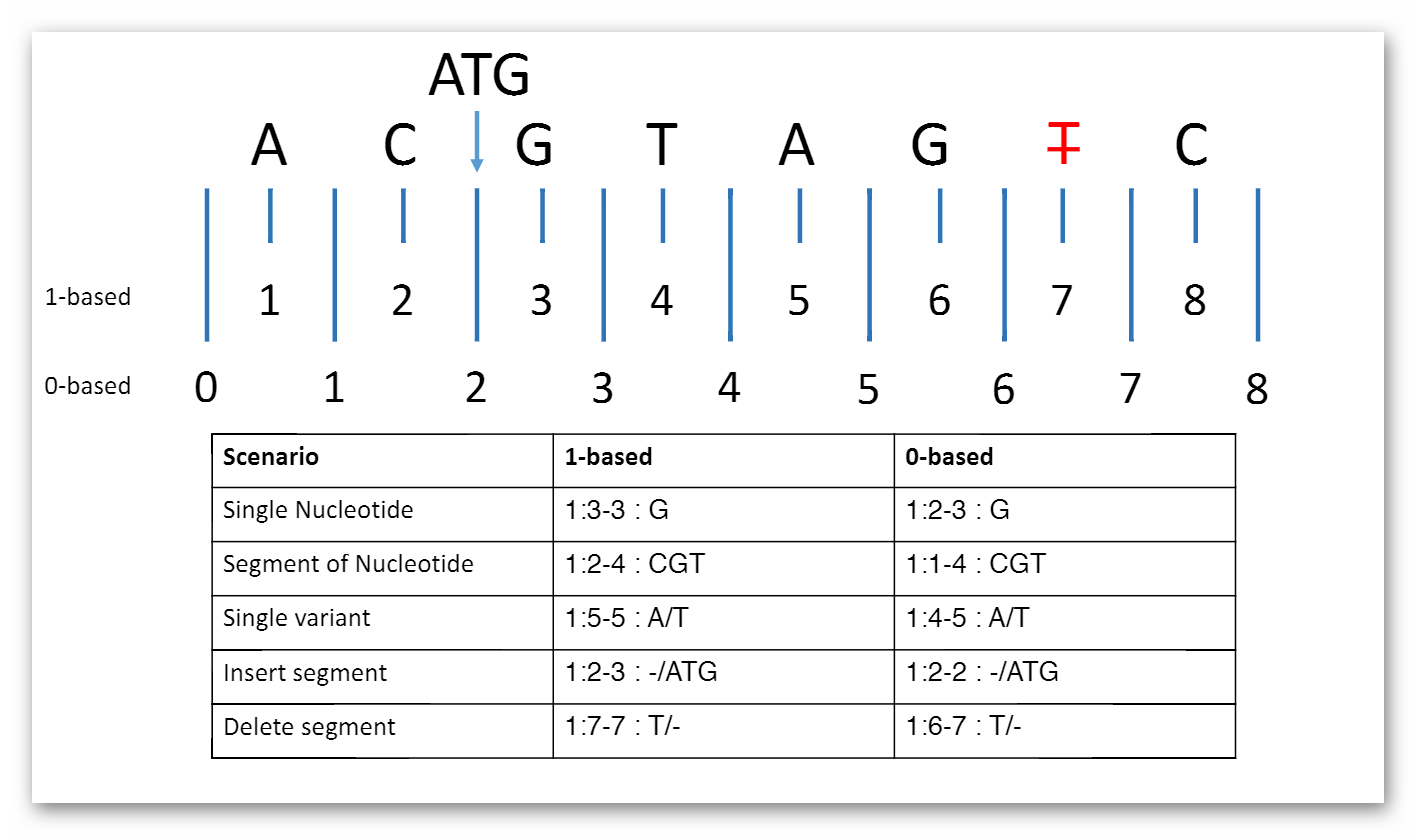
Here are two examples that clarify the usage in both cases (they represent same segment part):
MolecularSequence.referenceSeq: Four optional ways are provided to represent reference sequence in MolecularSequence resource:
The window selects a range from the reference sequence (or genome) that is used to define building block of a current sequence (e.g. MolecularSequence resource instance 1).
MolecularSequence.referenceSeq.strand: Only two possible values can be made by strand, +1 for plus strand while -1 for minus strand. Since the directionality of the sequence string might be represented in different word in different omics scenario, below are simple example of how to map other expressions into its correlated value:
| Map to +1 | Map to -1 |
|---|---|
| 5′-to-3′ direction | 3′-to-5′ direction |
| Watson | Crick |
| Sense | Antisense |
| Positive | Negative |
MolecularSequence.quality: Quality scores for bases in the sequence. It is intended to be compliant with emerging regulatory needs needs (eg: those found at PrecisionFDA ).
MolecularSequence.variant: This complex element is used for encoding sequence. When the information of reference sequence and variants are provided, the observed sequence will be derived.
MolecularSequence.patient: This element points to a Patient identifier to show that this sequence is related to the same patient.
MolecularSequence.specimen: A pointer to specimen identifier, if needed.
MolecularSequence.device: A pointer to Device identifier which is used for describing sequencing method (such as chip id, chip manufacturer etc.)
MolecularSequence.pointer: A pointer to a MolecularSequence instance for the next sequence block to build a sequence graph.
MolecularSequence.repository: This complex element is used to provide a clarifying structure, a base URL, and/or relevant IDs when referring to an external repository.
GA4GH Repository Example. If the MolecularSequence resource refers to a GA4GH repository for read info, references to a GA4GH full sequence dataset should conform to GA4GH data models and accessed via the GA4GH API. The URL of a GA4GH repository, ids of a GA4GH variant and read group are contained in the MolecularSequence resource. The URL of a GA4GH repository is an api_base of a GA4GH server that could be called for sequence data. The GA4GH variant set is a collection of call sets and the GA4GH call set is a collection of variant calls, typically for one sample. A variant call represents a determination of genotype with respect to that variant.
VariantSet definition: A VariantSet is a collection of variants and variant calls intended to be analyzed together.
CallSet definition: A CallSet is a collection of calls that were generated by the same analysis of the same sample.
A read group is a collection of reads produced by a sequencer. A read group set typically models reads corresponding to one sample, sequenced one way, and aligned one way. The API reference of Google Genomics is a GA4GH repository built by Google and provides details of the data models, such as the resource representations.
We provide a detailed example to show how sequence resource can be used to represent record of observed sequence by different method.
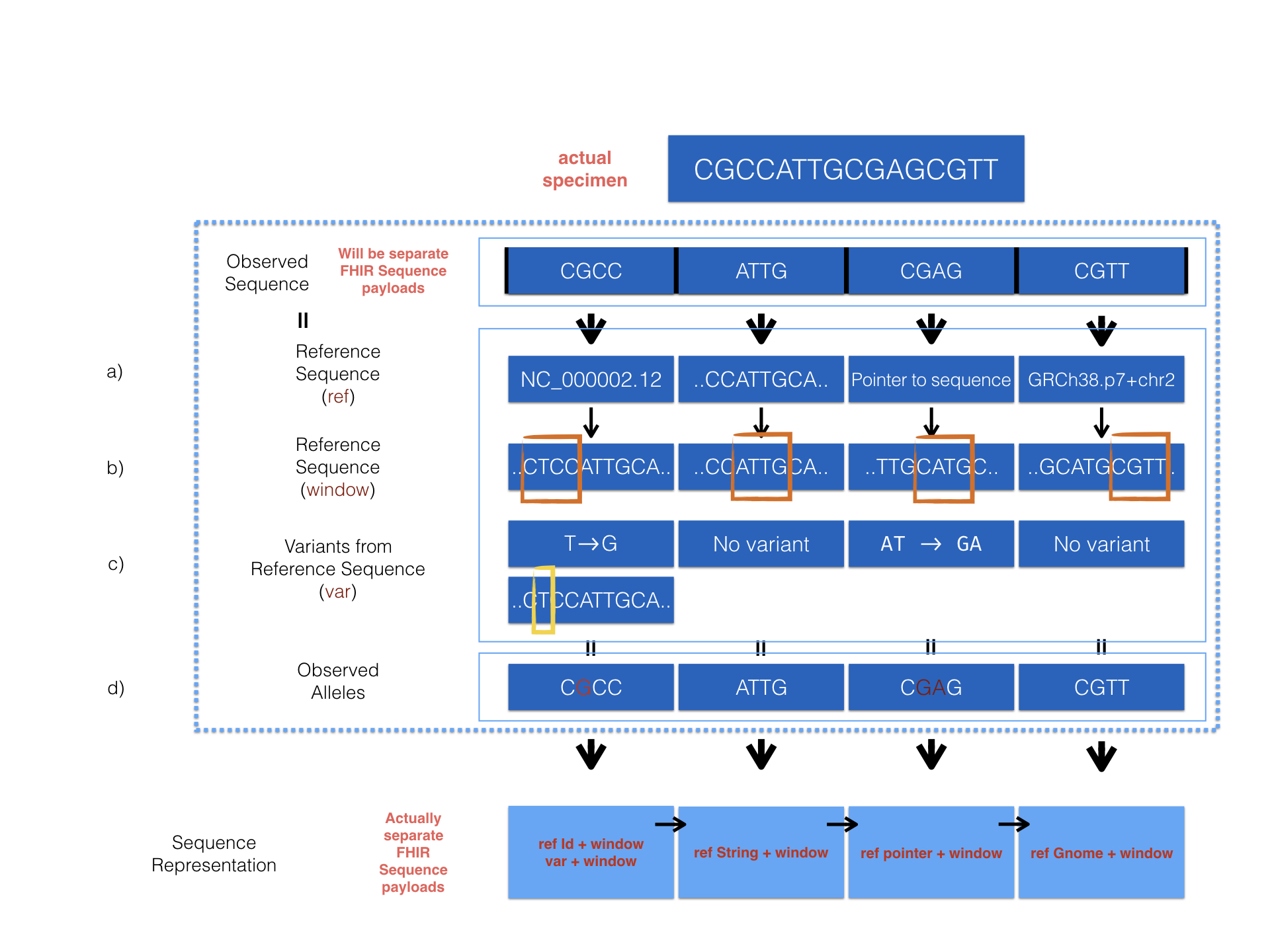
The diagram above describes 4 optional ways provided in the MolecularSequence resource to encode sequencing data. Here are the corresponding examples
We provide one example to show how precision FDA vcf data can be upload and comform FHIR specification, and how quality in sequence resource is represented by comparison between a reference sequence and the observed sequence.
We provide one example to show how a complex variant can be represented with the help of cigar. The deletion, insertion and mutation is represented in characters along with the number of repetition.
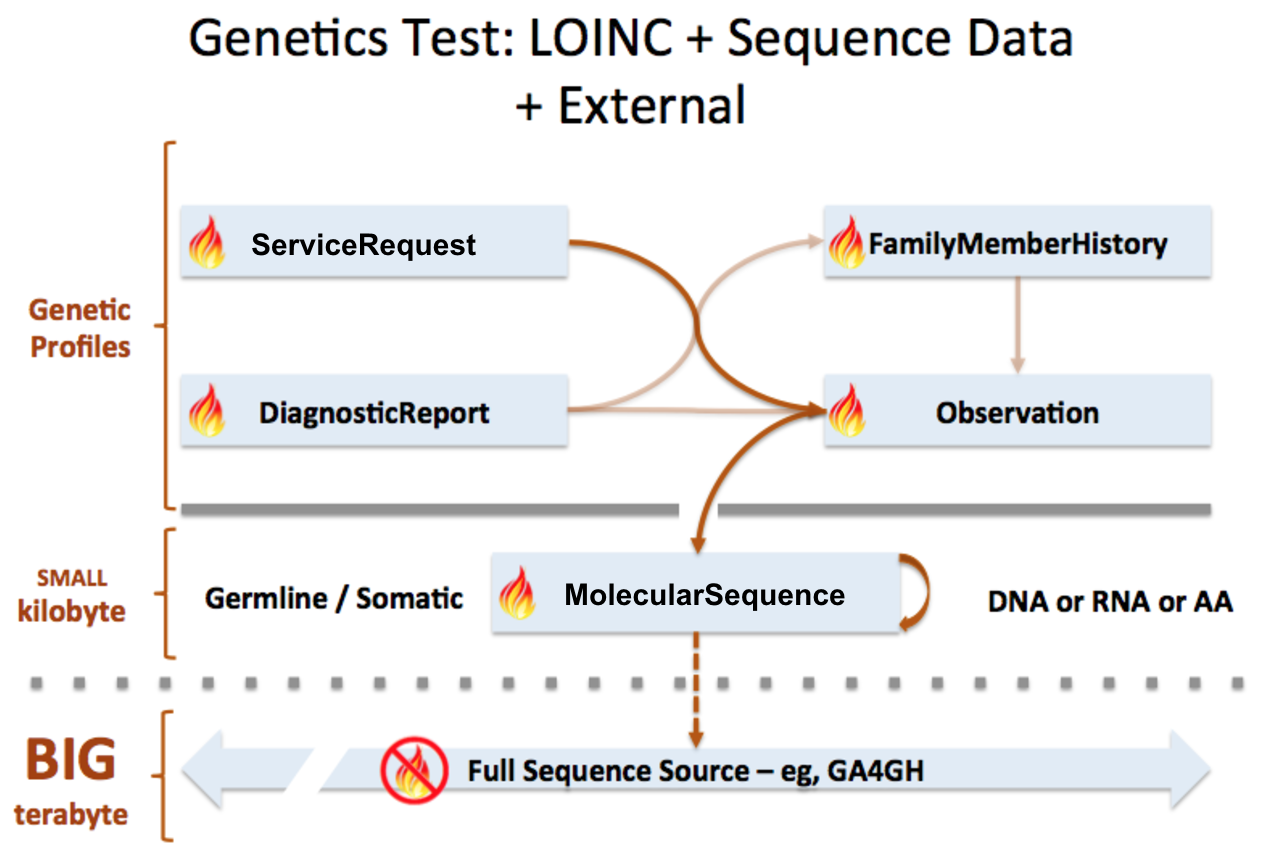
MolecularSequence resource is used to transmit and represent sequencing data. In FHIR Genomics, there are several genetics profiles containing internal pointers to MolecularSequence resource for genetic data reporting. Here is the summary:
MolecularSequence.pointer: A bundle of MolecularSequence instances can be connected by MolecularSequence.pointer to build sequence graph described in Section 3. MolecularSequence.pointer will point to the next sequence block.
MolecularSequence extension in Observation-genetics profile: Observation-genetics profile is used to report a genetic variant found in patients. MolecularSequence extension contains a pointer to MolecularSequence identifier which has related sequencing read info. It will provide reference/observed allele information and quality scores for each base/sequence block.
Observation-genetics based Observation resource is used for interpretative genetic data. MolecularSequence resource and genetics profiles will use internal pointers to Observation-genetics-profile based Observation instance for variant report.
MolecularSequence.observation: A pointer to genetics Observation instance which is used for interpretations of this sequence block. Interpretations are generally about genetic variant found in this sequence block.
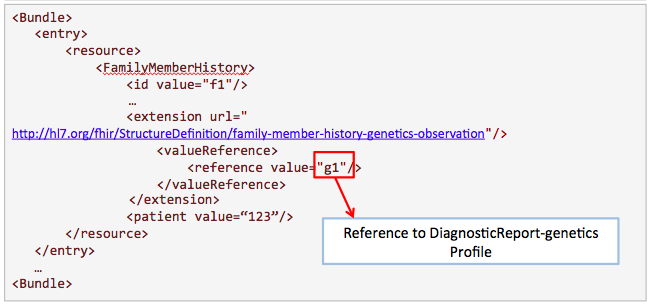
FamilyMemberHistory: A pointer to genetics Observation instance to report genetic test results of family member
During the development of the FHIR Genomics design, CGWG participants have commented on the earlier use cases and/or proposed new use cases. In this section, we list describe of these new cases and demonstrate how the proposed design will address them.
This example is proposed by Kevin Hughes. Family history is useful for clinicians to know more about the condition of the patient.
Get /FamilyMemberHistory?
_profile=http://hl7.org/fhir/StructureDefinition/familymemberhistory-genetic&
patient=123
The idea for a MolecularSequence resource grew out, in part, the SMART Platforms Project, which explored creating clinical genomic apps to integrated traditional EMR clinical data and genomic data to show data visualization and analysis, including CDS that depended upon both types of data. Below are a couple of examples. Several apps have already been designed including Genomics Advisor, SMART Precision Cancer Medicine, and Diabetes Bear EMR. Below, one of these apps will be described. To include other apps in this section, please feel free to add a note on it and how it uses FHIR/Genomics calls.
The SMART on FHIR Genomics Advisor was an app incorporating genomics data to show risk of disease, drug susceptibility, and related conditions based upon genotype. Technically, this app was architected (see below) by combining data from independent data services, a SMART on FHIR clinical server for clinical information and one for a SMART on FHIR Genomics data server for genomic data. The set of FHIR API calls that are necessary to support this app are shown below:
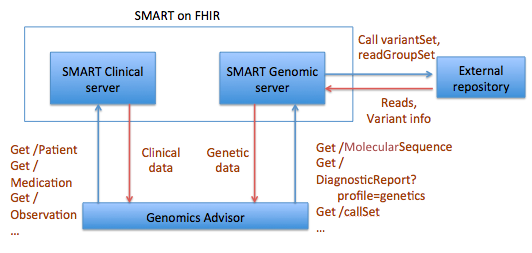
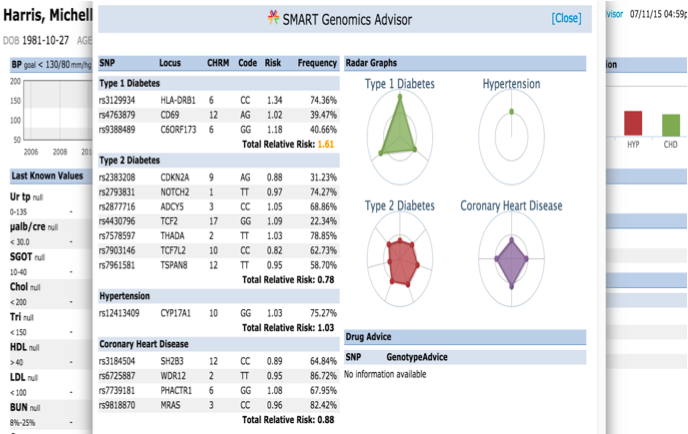
The presentation of the app looks like this:
FHIR ®© HL7.org 2011+. FHIR R5 Draft Ballot hl7.fhir.core#4.6.0 generated on Thu, Apr 15, 2021 12:30+1000.
Links: Search |
Version History |
Table of Contents |
QA Page |
Compare to R4 |
 |
Propose a change
|
Propose a change