
Pharmaceutical Quality (Industry)
1.0.0 - STU1
![]()
Pharmaceutical Quality (Industry)
1.0.0 - STU1
![]()
This page is part of the Pharmaceutical Quality (Industry) (v1.0.0: STU1) based on FHIR (HL7® FHIR® Standard) v5.0.0. This is the current published version in its permanent home (it will always be available at this URL). For a full list of available versions, see the Directory of published versions
| Official URL: http://hl7.org/fhir/uv/pharm-quality/ImplementationGuide/hl7.fhir.uv.pharm-quality | Version: 1.0.0 | |||
| Active as of 2024-05-08 | Computable Name: PharmaceuticalQualityIndustry | |||
| Other Identifiers: OID:2.16.840.1.113883.4.642.40.41 | ||||
This implementation guide defines a universal realm FHIR specification for the creation and exchange of structured pharmaceutical quality data internationally within or between biopharmaceutical companies and their stakeholders.
In-scope
Out of scope
Note:
This implementation guide is based upon FHIR version 5.0.0 and is a Universal Realm Specification.
This guide is primarily meant for but not necessarily limited to organizations involved in the following activities:
Pharmaceutical quality, also known as Chemistry, Manufacturing, and Controls (CMC) or just “Quality”, describes how a medicinal product is developed and manufactured; what suppliers and sites are involved; and the controls established to maintain the quality of the medicinal product over its lifecycle.
The scope, best practices, definitions, and content for pharmaceutical quality data are defined by the International Council for Harmonisation of Technical Requirements for Pharmaceuticals for Human Use (ICH), ICH Harmonised Tripartite Guideline: The Common Technical Document for the Registration of Pharmaceuticals for Human Use: Quality –M4Q(R1); Module 3: Quality, (12 September 2002), also known as the Common Technical Document (CTD).
The exchange and maintenance of pharmaceutical quality data account for the majority of all internal and external data interactions, internationally, within companies and between a company and stakeholders. Despite much of the content being sourced from internal systems that support structured data (e.g., pharmaceutical development systems, manufacturing site systems), and much of it being available for reuse across many medicinal products, pharmaceutical quality content has typically been authored, managed, and transmitted in unstructured Word files or PDFs. This is not sustainable, given the increasing volume, complexity, and frequency of quality work.
In an effort to adopt a more efficient operating model, the biopharmaceutical industry is about to transition into its third era or paradigm and, in addition to other benefits, this implementation guide will help accelerate this transition (see figure below). These eras are summarized as follows:
Figure: Advancement of the Submission and Decision-Making Ecosystems: The biopharmaceutical industry has undergone a previous transformation from paper-based submission to electronic paper submission. The industry is at the beginning of the next iteration of transformation, as signified by a transition to FHIR API data submission. This will be followed by further advancement as AI algorithms become increasingly prevalent. As the industry progresses in its journey towards digital maturity, faster submission and review processes will be made possible.
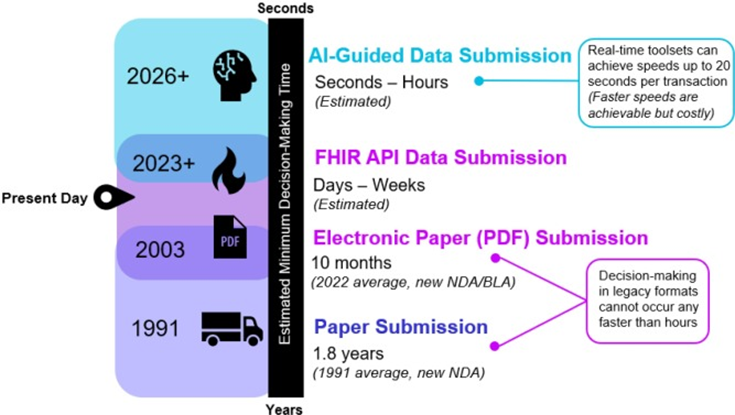 |
Currently, the end-to-end timescale for the pharmaceutical regulatory workflow is measured in months and years. This new paradigm, facilitated by this implementation guide, will use FHIR APIs and other supporting technologies to reduce the potential time for data exchange from months to days, hours, minutes, and eventually sub-seconds.
Establishing a viable, real-time method to exchange product quality information across biopharmaceutical manufacturing organizations can help to facilitate technology transfer, allow enhanced monitoring of facilities, and can potentially support developing advanced manufacturing methodologies, such as point-of-care manufacturing.
Importantly, spurring innovation in the biopharmaceutical sector is only one benefit of this real-time data exchange approach. Ultimately, the approaches highlighted within this implementation guide will help bring high quality medicinal products to market faster, and in a more cost-effective manner. This will also help create more robust manufacturing processes and more manageable global supply chains. This in turn will help reduce manufacturing down time, reduce the chance of shortages, and increase product quality and data compliance. Taken together, each of these benefits will enable speedier patient access to new treatments and minimize the post-approval life-cycle management burden for sponsors, while contributing favorably to overall global patient experience.
Given the Strategic Goals noted above, this implementation guide encourages the biopharmaceutical industry to adopt a new paradigm based on structured data with a high degree of granularity.
For example, each individual test (i.e., ObservationDefinition resources) is designed to be a discrete component. As a discrete component, this allows each test to be managed in a master data library where it can be centrally maintained, version controlled, and reused across different contexts or use cases. Each test, or ObservationDefinition, can be rolled up and combined with other resources into a Bundle that represents a larger concept like a stability study or test specification. It therefore becomes easier for sponsors to trace what tests, or what versions of tests, were used in any given stability studies or test specifications for any given batch over time. The same “rollup” or “bundling” approach is applied across all components used in the 14 data domains described in section 3 Domains Overview.
Once the individual components, like the ObservationDefinitions, are rolled up into bundles with other resources they can easily be exchanged between systems as FHIR compliant XML or JSON by default. However, if needed, these same bundles can be transformed into any other format that the biopharmaceutical sponsor may need. E.g., an XML or JSON bundle can be transformed into a PDF or Word document that maintains the traditional look and feal of monolithic ICH Common Technical Document (CTD) template.
This high degree of granularity maximizes a sponsor’s flexibility and choice in how they choose to manage their data; maximizes data reuse; reduces data redundancy; supports transformation into any desired output format; and makes this data more accessible for data science, ontologies, and artificial intelligence.
The following figure depicts (1) a potential future data pipeline for implementers; and (2) the intended scope of this IG’s use relative to the regulatory submission preparation workflow. As noted above, data structured according to this implementation guide is designed to be highly granular. The figure, and the following description, are provided to facilitate discussion and understanding of how to use the highly granular data models described in this implementation guide.
Figure: FHIR Data Pipeline for Pharmaceutical Quality (Industry)
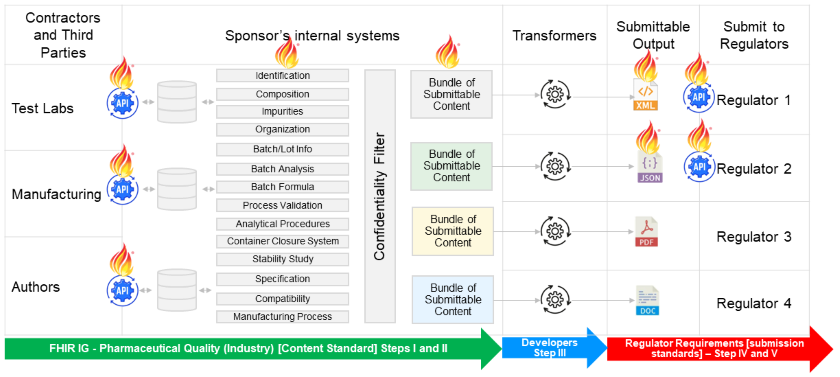 |
In summary, this potential future data pipeline could work as follows,
The scope of this implementation guide only covers data exchange activities for Step I, Step II, and Step III. The activities depicted in Step IV and Step V are out of scope for this implementation guide since they are determined by national regulations and regulatory guidance.
Biopharmaceutical industry:
Lantana Consulting Group:
This guide is developed and produced through the efforts of Health Level Seven International (HL7) as part of the Pharmaceutical Quality (Industry) project (Project ID 1800 and Project Scope Statement: PSS-2145).
The project team appreciates the support and sponsorship of the HL7 Public Health Work Group, and all volunteers and staff associated with the creation of this resource. The team appreciates the comments and input from the biopharmaceutical community as well as the HL7 volunteers who participate in the Biomedical Research and Regulation Work Group.
Health Level Seven, HL7, FHIR and the [FLAME DESIGN] are registered trademarks of Health Level Seven International, registered in the US Trademark Office. For acknowledgement of terminology content, please see the IP Statements section.
Please suggest improvements and provide notes about issues through HL7’s Jira Software (free account and log-in required).
HL7’s Confluence pages provide guidance on submitting Specifcation Feedback in Jira.
See also the HL7 FHIR Community Chat Zulip stream Pharm Quality Industry IG for discussion about the Pharmaceutical Quality (Industry) FHIR IG and BR&R Pharmaceutical Quality (PQ) - Industry Use Case project.
IG © HL7 International / Biomedical Research and Regulation. Package hl7.fhir.uv.pharm-quality#1.0.0 based on FHIR 5.0.0. Generated 2024-05-08
Links: Table of Contents |
QA Report
| Version History |
 |
Propose a change
|
Propose a change